Quantum reactive
scattering: Dynamics of Gas Phase
SN2 Reactions
Read our
invited review article: ChemPhysChem 5, 601 (2004)
SN2 reactions are nucleophilic bimolecular substitution
reactions. A nucleophilic species, e.g. Cl- attacks an
electrophilic centre, the carbon atom of the methyl group of , for example, a
CH3Br molecule. The C-Br bond breaks and a Cl-C bond is formed:
Cl- + CH3Br
--> ClCH3 + Br-
This mechanism plays an important role in Organic Chemistry. In freshman's
chemistry courses you learn that the reaction profile of an SN2
reaction shows a simple barrier. However, this is valid in the liquid phase
only. In the gas phase the profile looks completely different:
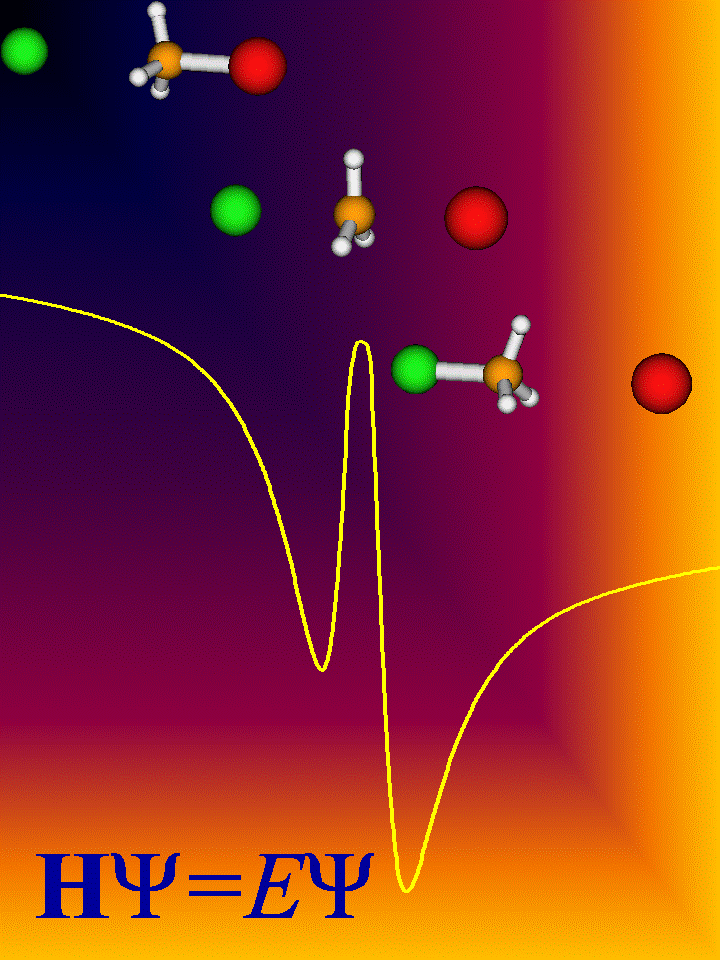
Due to the attraction between the charge of the nucleophile and the
dipole moment of the target molecule intermediate complexes are formed, one
before and one after passage of the transition state. The corresponding
potential wells are quite deep. It is possible that the reactive system is kept
in one of these complexes for a while. The complex can either dissociate back
to the educt side or surmount the barrier. In the second complex there are also
two ways to react. If one takes one of the complexes as starting point, the
reaction is essentially unimolecular. Being complex-forming bimolecular
reactions, gas phase SN2 reactions are on the cutting egde of
bi- and unimolecular reactions.
Because of the very special reaction profile the following questions
arise:
v
What is the influence of the
potential wells? How long are the lifetimes of the complexes? What can we say
about the wavefunctions of the corresponding resonance states?
v
What is the effect of excitation
of selected modes in the target species, e.g. the C-X stretching vibration or
the CH3 umbrella bending vibration?
v
What is the influence of
solvation on SN2 reactions?
v
How do different nucleophiles
and leaving groups the reaction rates?
In order to answer the above questions we first construct potential
energy hypersurfaces of the system which are based on results of high level ab
initio calculations (mainly coupled cluster calculations with relatively big
basis sets). We develop dimensionality reduced models in order to study the
influence of selected modes. The time-independent Schrödinger equation
is solved for continuum states by propagating (for a given total energy) the
wavefunction from the classically forbidden strong interaction region out to
the asymptotic region. The boundary conditions of the wavefunction yield -- via
the S matrix -- information about reaction probabilites, reaction
cross sections and rate constants. We also perform calculations in
order to obtain insight into the wavefunctions of the resonances. Using
the filter diagonalization method a lot of information can be obtained in
selected energy windows.